Würde das hier gerne mal alles bündeln da es bei diversen Erkrankungen die auch nicht ALS typisch sind aber neurologisch und die Muskeln betreffen, interessant ist. Es geht also um neurogene Muskelerkrankungen primär und myopatische die diese immitieren können.
Ich fange mal mit ALS Mimics an die ich so in den Publikationen fand, und dann kann man die Klinik näher erörtern und die Diagnostik. Bei der Klinik wäre insb interessant, ob es Charakteristika gibt und was im Vergleich zu einer ALS nit oder nur selten vorhanden ist, zB auch Faszikulationen, Spastiken usw.
Ich fange einmal mit den metabolischen an:
Metabolische ALS Mimics:
MR Bildgebungen zu den Krankheiten: Adrenoleukodystrophie
Befund:
Magnetresonanztomographie, zeigen eine symmetrische, flächige Degeneration der weißen Substanz mit Kontrastmittelaufnahme. Vorrangig betroffen sind dabei die Occipitallappen, der hintere Bereich des Corpus callosum, die Pyramiden- und die Hörbahn.[3][4]
Beispiel:
Diese T2-gewichtete MRT des Gehirns bei Adrenoleukodystrophie zeigt die Demyelinisierung im Marklager der hinteren Hirnabschnitte (hell), während das Marklager in den vorderen Abschnitten ein normales Signal aufweist (dunkel).
800px-Adrenoleukodystrophy.jpg
Weiteres Bild:
A.) Thorakal betonte Rückenmarkatrophie bei AMN (cMRT unauffällig)
B.) isolierte bilaterale Degeneration der Pyramidenbahnen bei AMN
C.) entzündliche Demyelisierung

Quelle: https://www.medmedia.at/klinik-ausga...ukodystrophie/
Ich fange mal mit ALS Mimics an die ich so in den Publikationen fand, und dann kann man die Klinik näher erörtern und die Diagnostik. Bei der Klinik wäre insb interessant, ob es Charakteristika gibt und was im Vergleich zu einer ALS nit oder nur selten vorhanden ist, zB auch Faszikulationen, Spastiken usw.
Ich fange einmal mit den metabolischen an:
Metabolische ALS Mimics:
| Adrenoleukodystrophie | Behandlung | Diagnostik | Unterschiede zur ALS |
| Gendefekt (meist Männer betreffend), Klinik: Tritt meist in der Kindheit auf, im Endstadium Demenz die zum Tod führt |
Antispastische Medikamente, Steroidhormone, Ursächlich: 4-Phenylbutyrat; Knochenmarktransplantationen | Diagnostik: Fettsäuren im Serum: Cerotinsäure (C26:0) und Quotienten C26:0/C24:0, C26:0/C22:0 und C24:0/C22:0 MRT: symmetrische, flächige Degeneration der weißen Substanz mit Kontrastmittelaufnahme; primär involviert: Occipitallappen; Corpus callosum, die Pyramiden- und die Hörbahn Gentest ABCD1 (weiter Diagnostik überflüssig? Sensitivität bekannt?) |
Früher Symptombeginn, Demenzielle Komponente (nicht FTD - Frontotemporale-Demenz), bei ausgeprägter Klinik bereits deutliche MRT Auffälligkeiten. |
Befund:
Magnetresonanztomographie, zeigen eine symmetrische, flächige Degeneration der weißen Substanz mit Kontrastmittelaufnahme. Vorrangig betroffen sind dabei die Occipitallappen, der hintere Bereich des Corpus callosum, die Pyramiden- und die Hörbahn.[3][4]
Beispiel:
Diese T2-gewichtete MRT des Gehirns bei Adrenoleukodystrophie zeigt die Demyelinisierung im Marklager der hinteren Hirnabschnitte (hell), während das Marklager in den vorderen Abschnitten ein normales Signal aufweist (dunkel).
800px-Adrenoleukodystrophy.jpg
Weiteres Bild:
A.) Thorakal betonte Rückenmarkatrophie bei AMN (cMRT unauffällig)
B.) isolierte bilaterale Degeneration der Pyramidenbahnen bei AMN
C.) entzündliche Demyelisierung
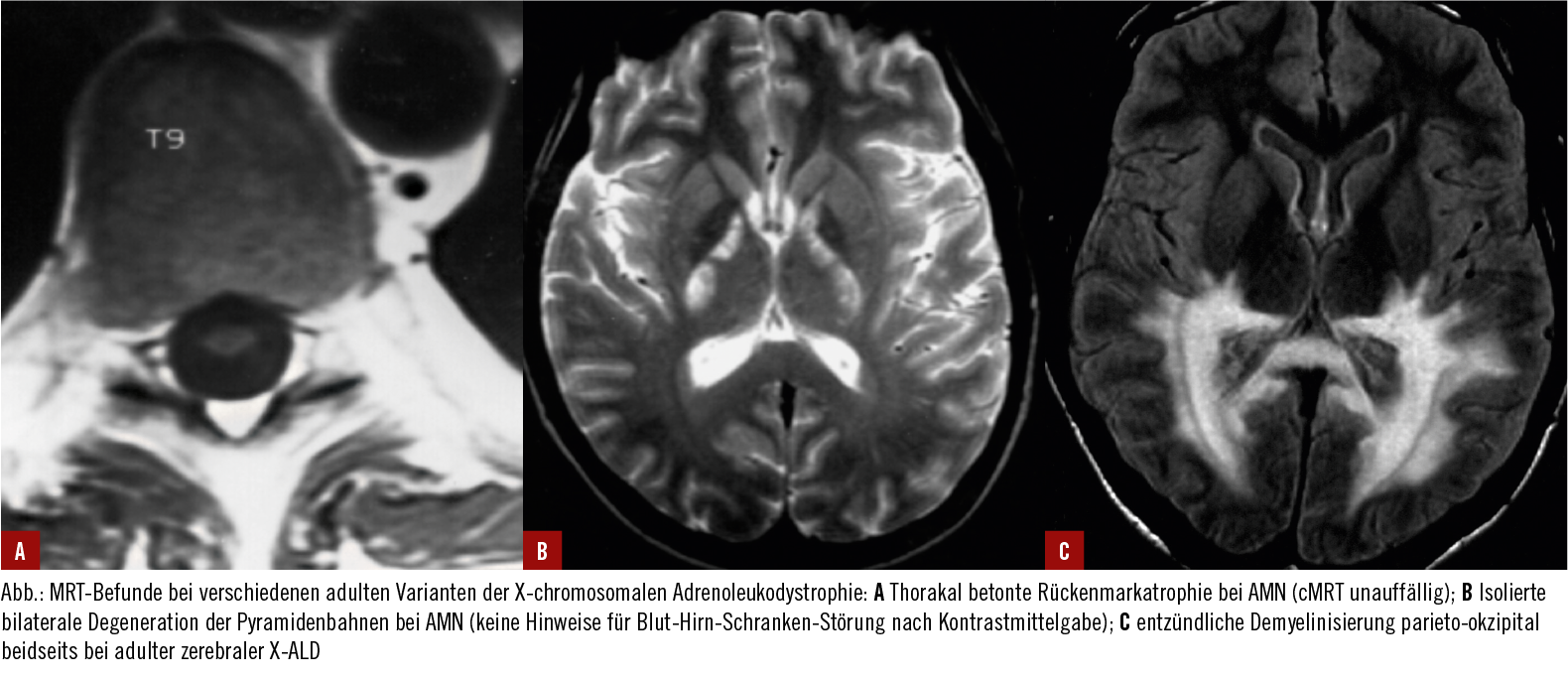
Quelle: https://www.medmedia.at/klinik-ausga...ukodystrophie/
Die X-chromosomal vererbte Adrenoleukodystrophie (X-ALD) ist die häufigste hereditäre Leukodystrophie und zählt pathophysiologisch zu den Peroxisomenerkrankungen.
Es liegt die Störung nur einer spezifischen Enzymfunktion bei morphologisch intakten Peroxisomen vor, Folge sind gestörter Abbau und Anreicherung überlangkettiger Fettsäuren (VLCFA).
Klinische Manifestation und Verlauf sind sehr heterogen; am häufigsten sind die kindlich-zerebrale Verlaufsform und die Adrenomyelopathie bei Erwachsenen.
Die Diagnose stützt sich auf biochemische (VLCFA) und molekulargenetische (ABCD-Gen) Bestimmungen, zerebrales MRT und evozierte Potenziale.
Behandlungsstrategien sind Stoffwechseltherapie und/oder hämatopoetische Stammzelltherapie bzw. Gentherapie.
Es liegt die Störung nur einer spezifischen Enzymfunktion bei morphologisch intakten Peroxisomen vor, Folge sind gestörter Abbau und Anreicherung überlangkettiger Fettsäuren (VLCFA).
Klinische Manifestation und Verlauf sind sehr heterogen; am häufigsten sind die kindlich-zerebrale Verlaufsform und die Adrenomyelopathie bei Erwachsenen.
Die Diagnose stützt sich auf biochemische (VLCFA) und molekulargenetische (ABCD-Gen) Bestimmungen, zerebrales MRT und evozierte Potenziale.
Behandlungsstrategien sind Stoffwechseltherapie und/oder hämatopoetische Stammzelltherapie bzw. Gentherapie.
Kommentar