Es wird ja zwischen 2 MN und "peripheren" 2 MN unterschieden. Wo ist da der Unterschied?
Das ist aber eigentlich Grundlagenwissen, welches man haben sollte, wenn man sich mit MNE beschäftigt...
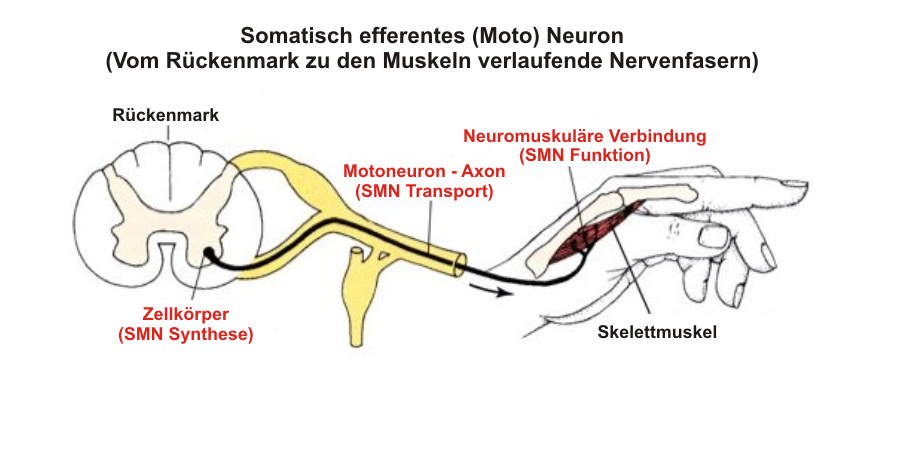
Funktionell werden die peripheren Nerven (also die Axone von den Nervenkernen ausgehend zum Endorgan/Muskel führend), Endplatten und Muskeln zum 2. MN gerechnet, daher manchmal auch peripheres MN genannt, obwohl das so formuliert nicht ganz korrekt ist.
Und auch wenn die Vorderhornzellen im Rückenmark, die Hirnnervenkerne im Gehirn liegen und damit anatomisch dem ZNS angehören, werden funktionell Läsionen der Vorderhornzellen und der Hirnnervenkerne selbst ebenso wie deren Axone als periphere Läsionen oder Läsionen des zweiten MN mit konsekutiver Muskelatrophie und Hypotonie bezeichnet - im Gegensatz zu den zentralen Läsionen im Motorkortex und der weißen Substanz des Rückemmarks und Gehirns (Pyramidenbahn) mit Reflexenthemmung und abnormer Steuerung der Muskeln.

Kommentar